




- -
- 100%
- +
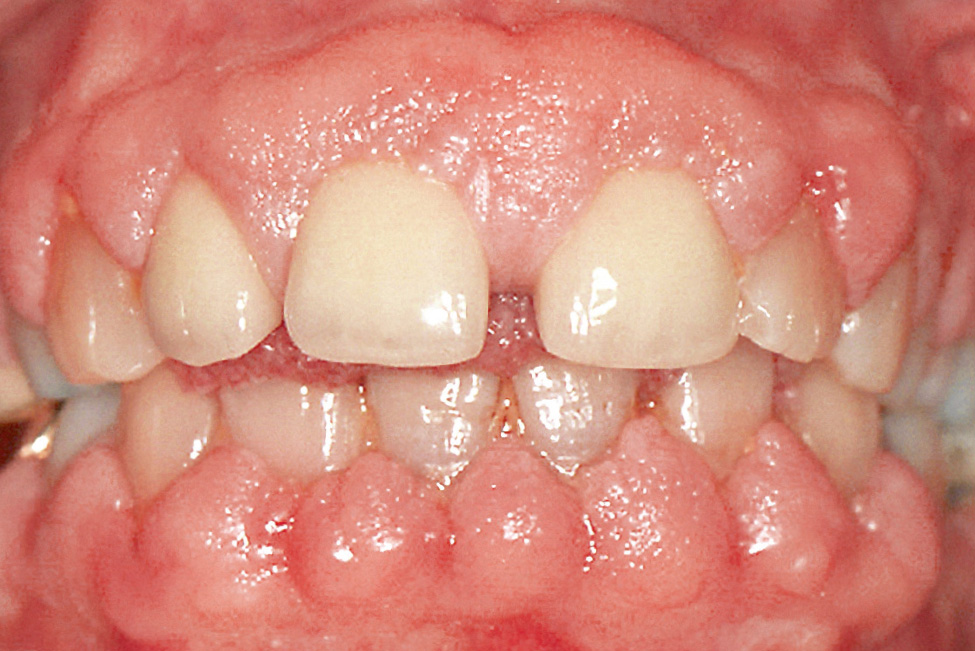
Abb. 4 Phenytoininduzierte Gingivawucherung.
Kalziumkanalblocker/-antagonisten
Kalziumkanalblocker werden hauptsächlich zur Therapie von pektangiösen Beschwerden und Bluthochdruck eingesetzt. Neben Verapamil (< 5 %)10, Amlodipin11, Diltiazem etc. wird vor allem Nifedipin8,9 aus der Gruppe der Kalziumkanalblocker mit Gingivawucherungen assoziiert (s. Tab. 2). Die Prävalenz von Gingivawucherungen nach Einnahme von Kalziumkanalblockern ist sehr unterschiedlich und ist mit bis zu 20 % bei Nifedipin und Diltiazem am höchsten.
Die Gingivavergrößerung ähnelt klinisch und histologisch der hydantoininduzierten Wucherung und manifestiert sich am ausgeprägtesten im Bereich der labialen Papillen (Abb. 5). Die Veränderungen bilden sich nach Absetzen des Medikaments häufig wieder zurück. Durch eine regelmäßige Entfernung der Plaque ist es möglich, das Ausmaß der Wucherung deutlich zu reduzieren und oft auch vollständig zu eliminieren.
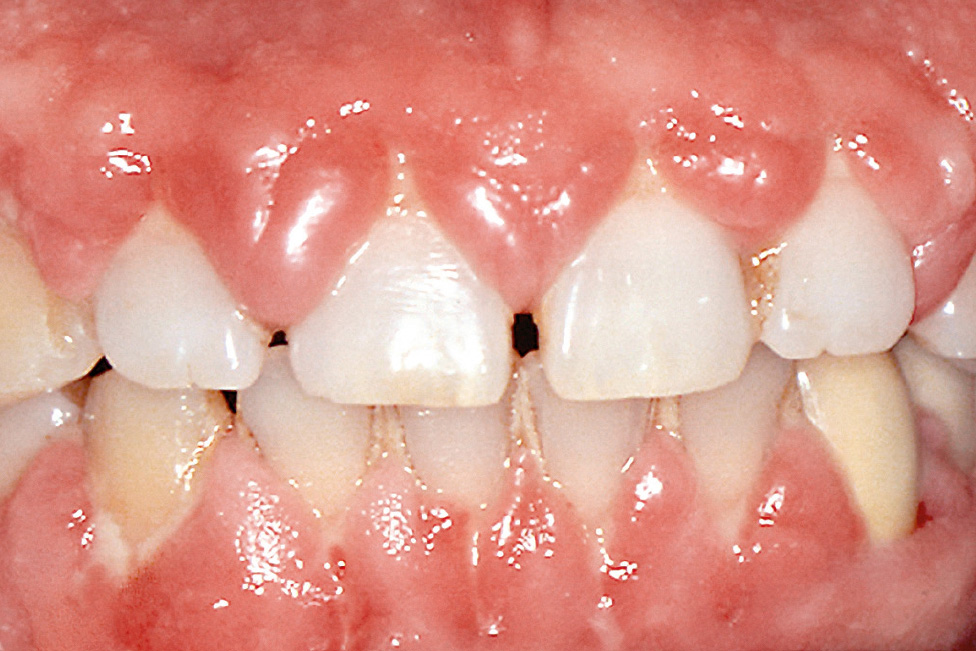
Abb. 5 Gingivawucherung bei Medikation mit einem Kalziumkanalblocker.
Nicht plaqueinduzierte Gingivavergrößerungen (vererbte Gingivafibrose)
Die nicht plaqueinduzierten Gingivavergrößerungen sind primär nicht bakterieller Genese. Durch Ausbildung von Pseudotaschen und die dadurch erschwerte persönliche Mundhygiene kann es aber sekundär zu entzündlichen Überlagerungen kommen.
Idiopathische Gingivawucherungen können einen genetischen Ursprung haben; die genaue Ätiologie ist bisher allerdings nicht geklärt. Die Erkrankung kann autosomal dominant vererbt werden, der pathogenetische Zusammenhang ist bisher aber nicht bekannt. Die Gingivawucherung kann als alleiniges Symptom auftreten oder in Verbindung mit anderen pathologischen Erscheinungen (z. B. Hypertrichiose). Die gutartige Wucherung besteht meist aus einem dichten, faserreichen (Kollagen), aber zellarmen Bindegewebe (Abb. 6). Treten die Wucherungen symmetrisch, z. B. retromolar im Ober- und/oder Unterkiefer auf, werden sie oft auch als symmetrische, periphere Fibrome bezeichnet.
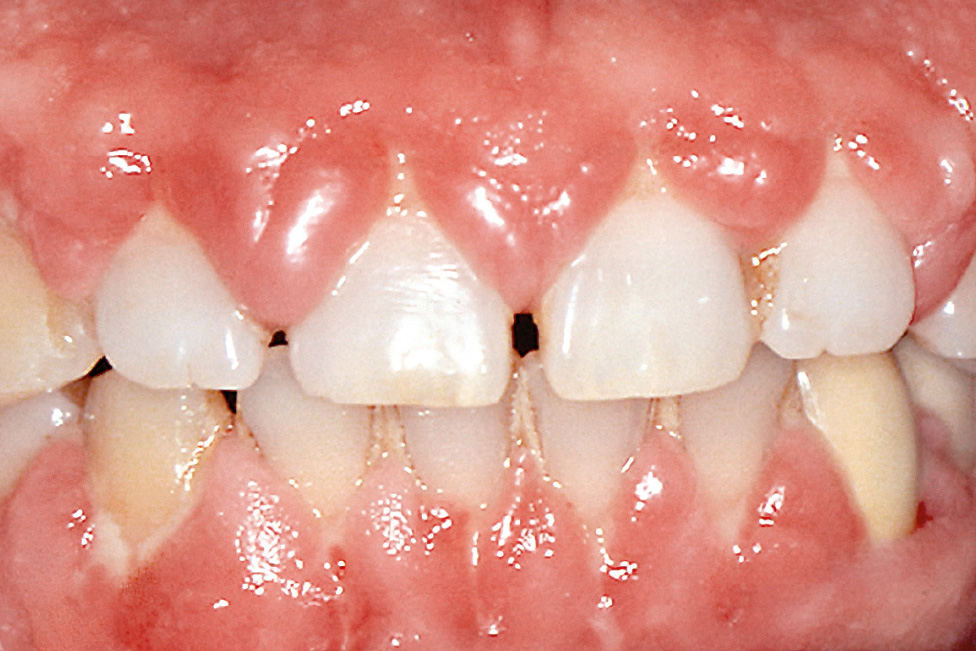
Abb. 6 Hereditäre Gingivawucherung.
Die klinischen Zeichen der Erkrankung können von Geburt an erscheinen, zeigen sich aber meist erst mit Durchbruch der Milch- bzw. bleibenden Zähne. Eine spontane Remission der fibrösen Verdickung tritt nicht auf. Falls es durch das überschüssige Gewebe zu Tascheninfektionen, funktionellen Störungen oder Behinderung im Zahndurchbruch kommt, muss die Wucherung operativ entfernt werden. Nach der chirurgischen Exzision kommt es allerdings häufig zu Rezidiven.
Literatur
1. Caton JC, Armitage G, Berglundh T et al. A new classification scheme for periodontal and peri-implant diseases and conditions – Introduction and key changes from the 1999 classification. J Clin Periodontol 2018;45(Suppl 20):S1–S8.
2. Murakami S, Mealey BL, Mariotti A et al. Dental plaque–induced gingival conditions. J Clin Periodontol 2018; 45(Suppl 20):S17–S27.
3. Chapple ILC, Mealey BL, Van Dyke TE et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol 2018;45(Suppl 20):S68–S77.
4. Sills ES, Zegarelli DJ, Hoschander MM et al. Clinical diagnosis and management of hormonally responsive oral pregnancy tumor (pyogenic granuloma). J Reprod Med 1996;41:467–470.
5. Ziskin DE, Nesse GJ. Pregnancy gingivitis. history, classification, etiology. Am J Orthod Oral Surg 1946;32:390–432.
6. Dongari-Bagtzoglou A. Drug-associated gingival enlargement. J Periodontol 2004;75:1424–1431.
7. Angelopoulous AP, Goaz PW. Incidence of diphenylhydantoin gingival hyperplasia. Oral Surg Oral Med Oral Pathol 1972;34:898–906.
8. Barclay S, Thomason JM, Idle JR et al. The incidence and severity of nifedipine-induced gingival overgrowth. J Clin Periodontol 1992;19:311–314.
9. Nery EB, Edson RG, Lee KK et al. Prevalence of nifedipine-induced gingival hyperplasia. J Periodontol 1995;66:572–578.
10. Miller CS, Damm DD. Incidence of verapamil-induced gingival hyperplasia in a dental population. J Periodontol 1992;63:453–456.
11. Jorgensen MG. Prevalence of amlodipine-related gingival hyperplasia. J Periodontol 1997;68:676–678.
12. Hassel TM, Hefti AF. Drug-induced gingival overgrowth: old problem, new problem. Crit Rev Oral Biol Med 1991;2:103–137.
Peter Eickholz, Filip Klein, Katrin Nickles
Parodontitis als Symptom von Syndromerkrankungen6Einleitung
Einige genetisch bedingte Syndromerkrankungen gehen mit früh beginnenden und sehr rasch verlaufenden Parodontitiden einher. Diese Erkrankungen werden in der Klassifikation von 2018 als systemische Erkrankung mit wesentlichem Einfluss auf parodontalen Attachmentverlust durch Einfluss auf parodontale Entzündung (z. B. Papillon-LefèvreSyndrom) oder als systemische Erkrankungen, die unabhängig von Parodontitis parodontalen Attachmentverlust verursachen können (z. B. Langerhans-Zell-Histiozytose), berücksichtigt1,2. In erster Linie gehören dazu angeborene Defekte des zellulären und/oder humoralen Immunsystems sowie andere Erkrankungen, die mit einer gestörten Leukozytenfunktion verbunden sind. Seltener sind frühzeitig beginnende Parodontitiden die Folge von Defekten des parodontalen Bindegewebes. Viele dieser Erkrankungen gehören zu den seltenen Erkrankungen („orphan diseases“) mit einer Prävalenz < 1:2.0003.
Syndrom
Ein Syndrom ist ein Muster multipler Anomalien bzw. eine Gruppe von Krankheitszeichen, deren ursächliche Verbindung bekannt ist oder vermutet wird. Im weiteren Sinne ist es ein sich stets mit etwa dem gleichen Symptomenkomplex manifestierendes Krankheitsbild mit unbekannter, uneinheitlicher oder multifaktorieller Ätiologie bzw. Pathogenese.
Funktionsstörungen der neutrophilen Granulozyten
Leukozyten-Adhäsionsdefekt-Syndrome
Die verschiedenen Formen des Leukozyten-Adhäsionsdefekt-Syndroms (LAD) sind sehr seltene angeborene, autosomal-rezessiv vererbte primäre Immundefekte (Orpha-Kennnummer: ORPHA2968/ICD-10-Code: D72.0). Das vorherrschende klinische Kennzeichen sind wiederkehrende bakterielle Infektionen mit oft schwerem, lebensbedrohlichem Verlauf, die schwer zu therapieren sind. Es kommt zu früh beginnender und sehr rasch verlaufender Parodontitis, die mit dem Durchbruch der Milchzähne einsetzt, sämtliche Zähne der ersten und zweiten Dentition befällt und durch eine heftige Entzündungsreaktion der Gingiva mit brombeerartiger feuerroter Schwellung charakterisiert ist. Vorzeitiger Zahnverlust kann durch parodontale Therapie bisher nicht verhindert werden. Sie werden durch eine defekte Adhäsion der Leukozyten an aktivierte Endothelien hervorgerufen, die die Migration der Leukozyten in mikrobiell kontaminierte oder besiedelte Gewebe verhindert. Bei Infektionen kommt es zu einem dramatischen Anstieg der myeloischen Leukozyten im Blut, die aber nicht an ihren Einsatzort gelangen können.
Bei LAD-I liegt keine oder eine reduzierte Expression der β2-Integrin-Untereinheit vor (β2Integrine: αLβ2 [CD11a/CD18, LFA-1], αMβ2 [CD11b/CD18, MAC-I, CR3], αXβ2 [CD11c/CD18, p150/95], αDβ2 [CD11d/CD18]), was zu einem Defekt der engen Adhäsion der Leukozyten führt. Bisher wurden über 80 Mutationen gefunden. Es lassen sich schwere (< 2 % Expression) von moderaten (2–30 %) Formen unterscheiden. Patienten mit moderatem LAD-I können die Kindheit mit Antibiotika überleben. Es kommt zu einer verzögerten Ablösung der Nabelschnur und dramatisch verzögerter bzw. dysplastischer Heilung infizierter Wunden. β2-Integrine haben auch eine Aufgabe in der Interaktion von neutrophilen Granulozyten (PMNs) und Makrophagen. Die reduzierte Expression kann so die Beseitigung apoptotischer PMNs aus infizierten Wunden beeinträchtigen.
Bei LAD-II besteht ein Defekt in der Fucosylation von Oligosacchariden einschließlich SialylLewis X, das sich auf P-Selektin-Gykoprotein-1 befindet. Dieser Defekt führt zu einem gestörten „rolling“ der Leukozyten auf aktiviertem Endothel. Patienten mit LAD-II sind zumeist geistig retardiert, von kleiner Statur und weisen einen abnormalen Erythrozyten-Phänotyp (Bombay-Phänotyp) auf.
Patienten mit LAD-III zeigen ähnliche klinische Zeichen wie bei LAD-I mit einer defizienten engen Adhäsion der Leukozyten an aktivierte Endothelien. Im Unterschied zu LAD-I werden die β2-Integrine aber in (nahezu) normalen Mengen exprimiert. Es scheint eine Störung beim „inside-out signalling“ zu bestehen, das die Integrine von einem inaktiven Zustand niedriger Affinität in einen aktivierten Zustand überführt, der eine Bindung an Liganden erlaubt (Aktivierung). Darüber hinaus bestehen eine Blutungsneigung sowie eine Störung der Aktivierung von Integrin αIIbβ3, das wesentlich die Aggregation und Adhäsion der Thrombozyten vermittelt.
Prognose: Bei intensiver Antibiotikatherapie wird das Erwachsenenalter erreicht. Knochenmarkstransplantationen waren bei einigen Patienten erfolgreich4.
Papillon-Lefèvre-Syndrom
Das Papillon-Lefèvre-Syndrom (PLS; Keratosis palmoplantaris – Periodontopathie; ORPHA678/ICD-10: Q82.8) beschreibt seltene, autosomal-rezessiv erbliche Mutationen des Kathepsin-C-Gens, die zur Inaktivität dieser Cysteinprotease führen. Kathepsin C aktiviert die Serinproteasen (Elastase, Kathepsin G, Protease 3, Neutrophilen-Serinprotease 4) der neutrophilen Granulozyten. Damit ist die erste Linie der parodontalen Infektabwehr beeinträchtigt. Die charakteristischen Symptome sind palmoplantare Hyperkeratosen mit einer bereits im Milchgebiss einsetzenden und sehr rasch verlaufenden Parodontitis (Abb. 1a bis d). Allerdings sind auch atypische Verläufe bekannt, bei denen entweder nur Hautsymptome oder nur die im Milchgebiss einsetzende und rasch verlaufende Parodontitis auftreten6. Die Häufigkeit beträgt 1–4:1.000.000. Untersuchungen der neutrophilen Granulozyten haben in manchen Fällen gestörte Zellfunktionen wie eine verminderte Motilität, Chemotaxis und Phagozytose sowie eine verminderte Produktion von Sauerstoffradikalen gezeigt, die sich in manchen Fällen nach der therapeutischen Intervention normalisierten. In der subgingivalen Plaque werden zumeist vermehrt anaerobe gramnegative Keime und vor allem Aggregatibacter actinomycetemcomitans nachgewiesen.
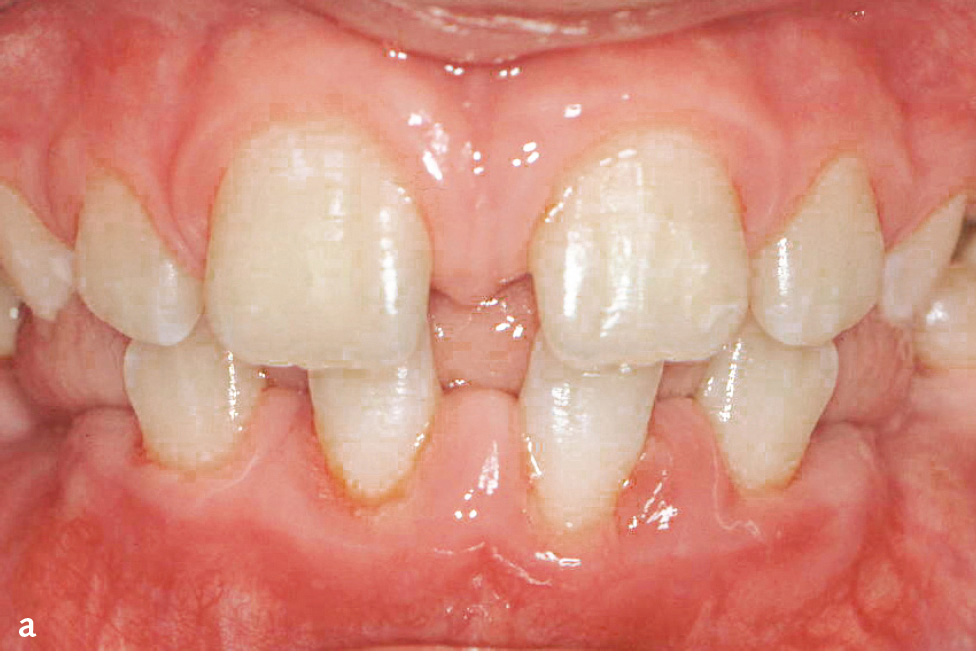
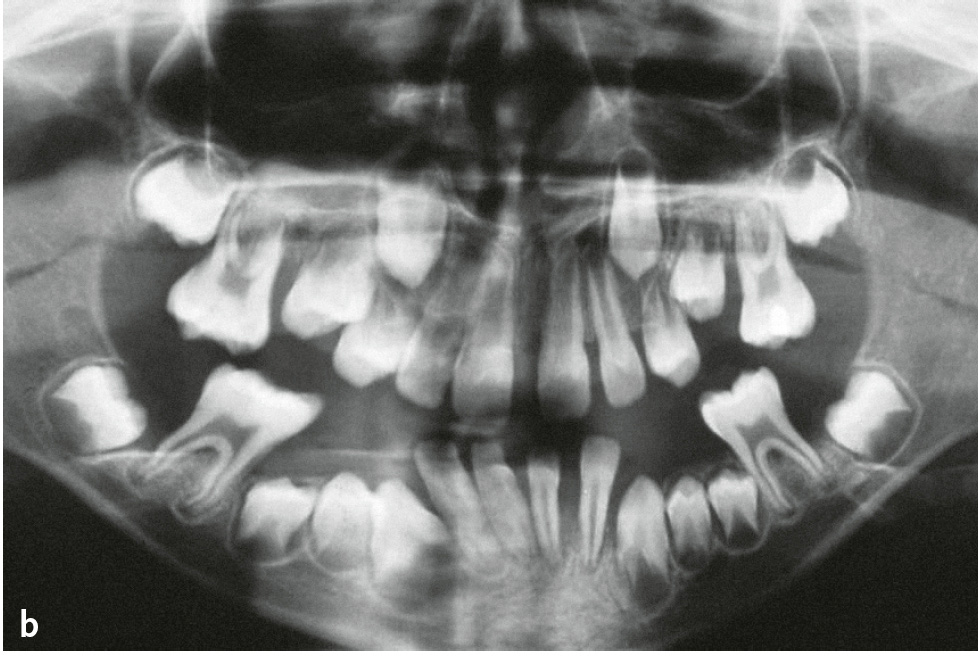
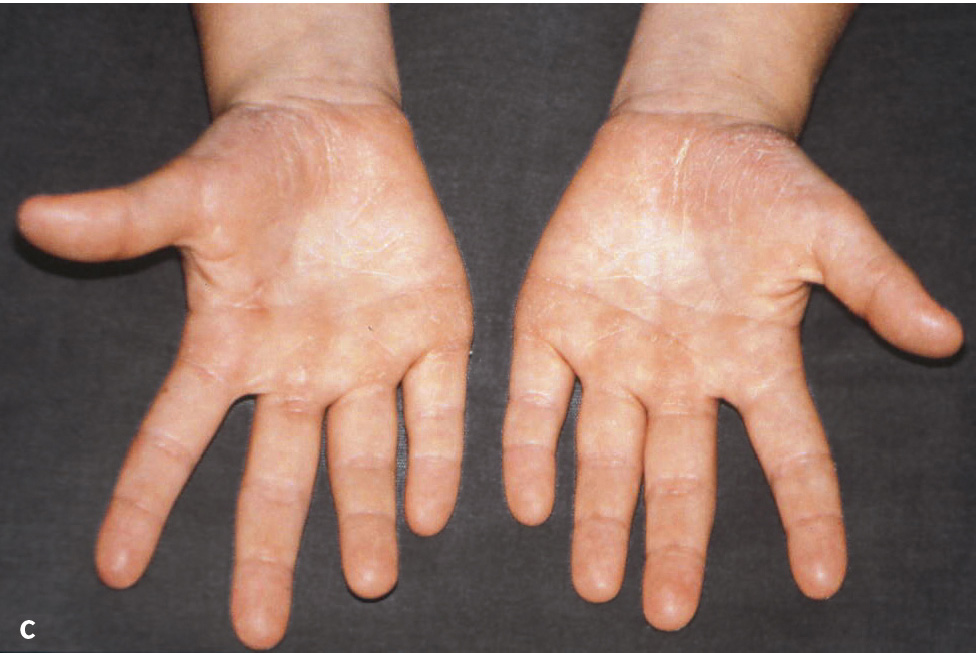
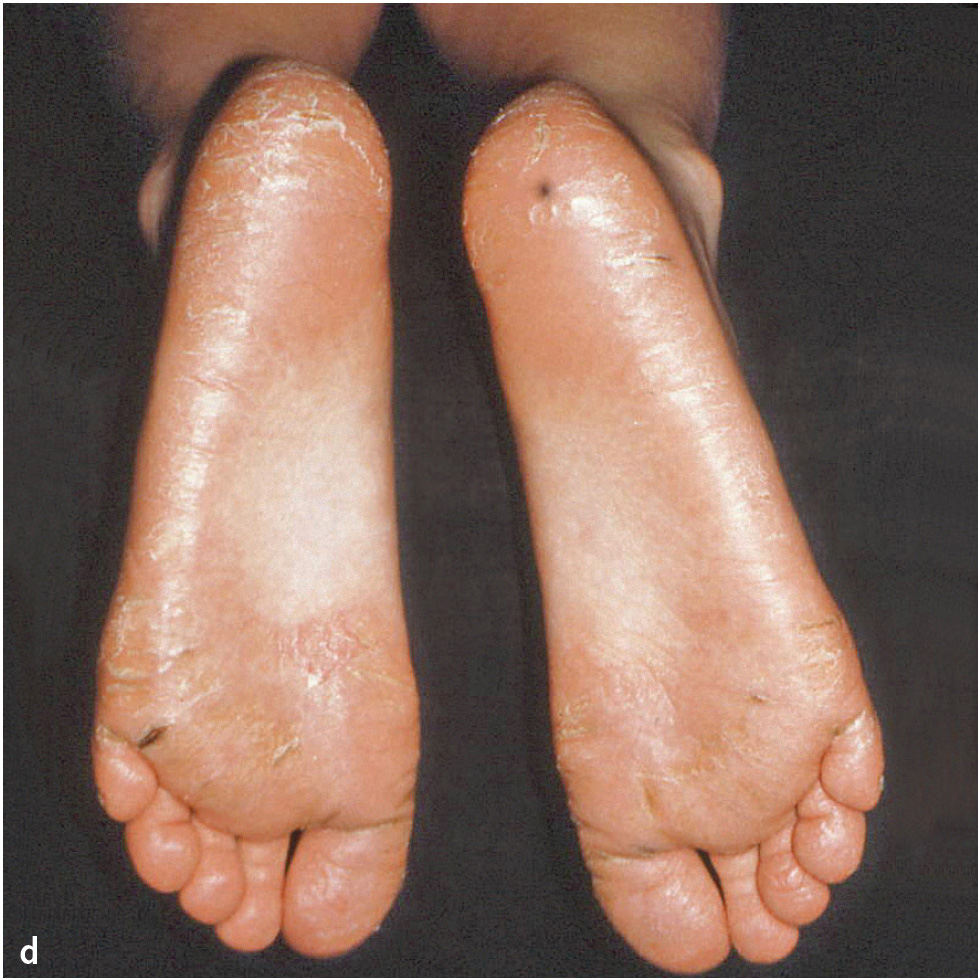
Abb. 1a bis d Männlicher Patient mit Papillon-Lefèvre-Syndrom: a) klinische Ansicht im Alter von 7 Jahren und 9 Monaten (vor Therapie: ausgeprägte Mobilität der Unterkieferschneidezähne, Sondierungstiefen an ersten Molaren und Unterkieferschneidezähnen von 9 bis 15 mm); b) Panoramaschichtaufnahme; Hyperkeratosen an c) Hand- und d) Fußflächen (Abbildungen aus Kugel et al. 20015).
Die Symptomatik umfasst über die palmoplantaren Hyperkeratosen und die im Milchgebiss einsetzende und rasch verlaufende Parodontitis hinaus psoriasiforme Hyperkeratosen an Ellenbogen und Knien, Nageldystrophie, Mineralisationen der Dura mater und gehäufte bakterielle Infekte.
Die Therapie der Hautveränderungen erfolgt zum Teil mit oralen Retinoiden, aber auch urathaltigen Salben. Eine erfolgreiche Therapie der Parodontitis ist möglich. Der Erfolg scheint von verschiedenen Faktoren abzuhängen: möglichst frühzeitiger Beginn (ggf. schon im Milchgebiss), Elimination von A. actinomycetemcomitans und engmaschige professionelle Nachsorge (Abb. 1e und f)7.
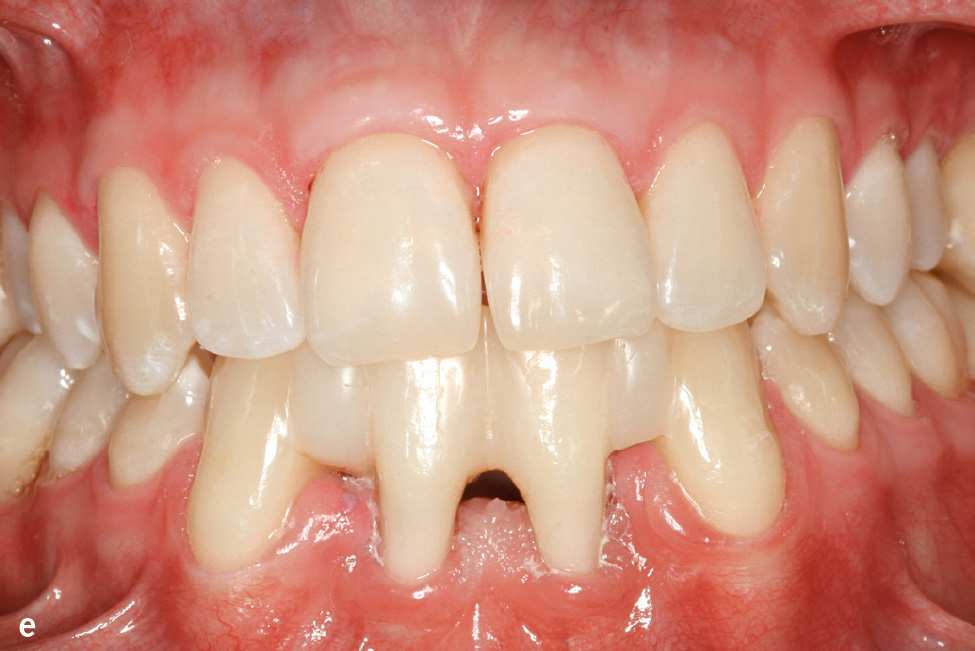
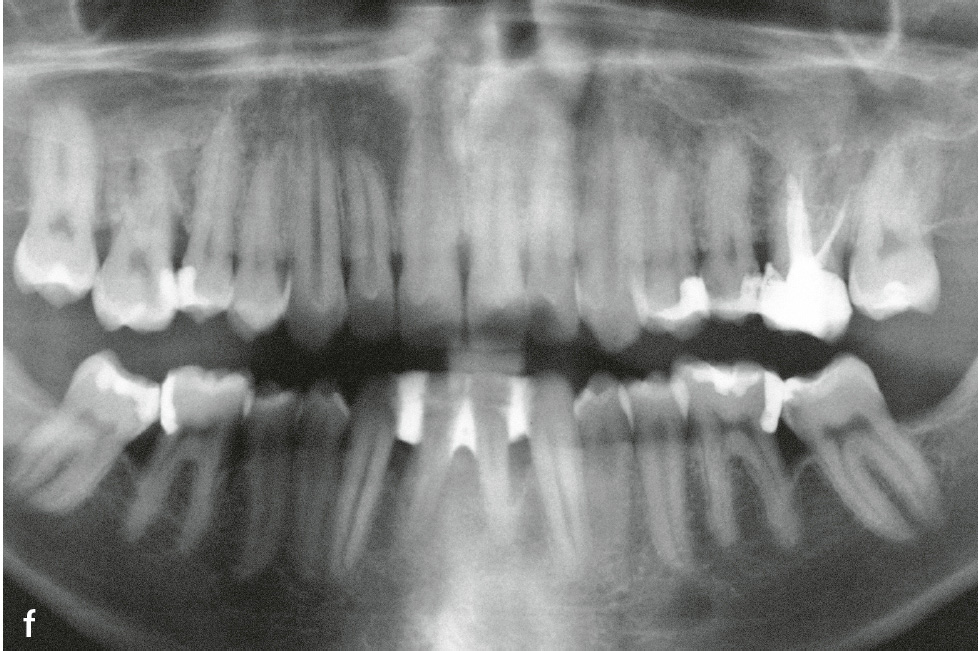
Abb.1e und f e) Klinische Ansicht im Alter von 22 Jahren; 14 Jahre nach nichtchirurgischer antiinfektiöser Therapie mit unterstützender Gabe von Amoxicillin und Metronidazol und anschließender unterstützender Parodontitistherapie anfänglich alle 2, später alle 3 Monate; f) Panoramaschichtaufnahme zu Abb. 1e.
Haim-Munk-Syndrom
Das Haim-Munk-Syndrom (HMS; Keratosis palmoplantaris – Parodontopathie – Onychogryphosis, Keratosis palmoplantaris diffusa mit Periodontopathie, Palmoplantarhyperkeratose – Parodontopathie – Onychogryphosis; ORPHA2342/ICD-10: Q82.8) ist ein weiteres autosomal-rezessiv vererbbares Syndrom, das bisher in einer jüdischen Bevölkerungsgruppe in Indien und Israel beschrieben wurde. Mit palmoplantaren Hyperkeratosen und früh beginnender sowie sehr rasch verlaufender Parodontitis ist es dem Papillon-LefèvreSyndrom ähnlich. Es zeigen sich allele Mutationen des Kathepsin-C-Gens. Weitere Symptome sind Hypertrophie und Krümmung der Nägel (Onychogryphose), Plattfüße (pes planus), Arachnodaktylie und Osteolyse der distalen Phalangen der Finger sowie Zehen (Akroosteolyse). Die Unterscheidung zwischen Papillon-Lefèvre- und Haim-Munk-Syndrom ist schwierig und setzt die Berücksichtigung der skelettalen Symptome von HMS voraus8.
Chediak-Higashi-Syndrom
Das Chediak-Higashi-Syndrom (CHS; ORPHA167/ ICD-10: E70.3) ist eine sehr seltene, autosomalrezessiv erbliche Stoffwechselanomalie, die durch Immundefekte, neurologische Dysfunktion (Muskelschwäche, Ataxie, Verlust der Sensorik, Nystagmus), Albinismus der Augen und der Haut sowie Blutungsneigung aufgrund einer Thrombozytenfunktionsstörung gekennzeichnet ist. Dem CHS liegen Mutationen des LYST(lysosomal trafficking regulator)-Gens zugrunde, das ein Protein kodiert, das in die Regulation der Lysosomen eingebunden ist. Typisch sind sehr große Einschlusskörper in nahezu allen granulierten Zellen (z. B. Granulozyten, Histiozyten, Mastzellen, Thrombozyten, Melanozyten, Schwann-Zellen, Neurone), die durch Fusion zytoplasmatischer Granula entstehen. Im Falle der Myelozyten erfolgt dies in frühen Reifungsstadien und führt zum Absterben von myeloischen Vorläuferzellen und damit zu moderater Neutropenie9.
Als Symptome zeigen sich mit Manifestation im Kindesalter eine Disposition zu rezidivierenden Infektionen, eine allgemeine Hypopigmentation, ein partieller Albinismus und ein Albinismus fundi occuli, eine Hepatosplenomegalie und eine Lymphadenopathie. Typischerweise geht diese Erkrankung mit einer früh beginnenden und sehr rasch verlaufenden Parodontitis einher.
Bei der Diagnostik zeigen sich im peripheren Blutbild eine Granulationsanomalie der Granulozyten und Lymphozyten (Riesengranula) und plasmatische Einschlusskörperchen in den myeloischen Zellen im Knochenmark. Pränatal sind eine fetale Blutuntersuchung und eine Haut-Haar-Biopsie möglich.
Die Therapie besteht in einer Knochenmarkstransplantation. Die Prognose ist aufgrund der Disposition zu septischen Prozessen im Kindesalter ungünstig. Die meisten Kinder sterben vor Erreichen des zehnten Lebensjahres an Infekten oder malignen Tumoren.
Kostmann-Syndrom
Das Kostmann-Syndrom (Infantile genetische Agranulozytose; ORPHA99749/ICD-10: D70.0) ist die häufigste angeborene, sich früh manifestierende, hochgradige Granulozytenfunktionsstörung mit Auftreten akuter, lebensbedrohlicher bakterieller (Staphylococcus aureus, Klebsiellen) und mykotischer (Aspergillus) Infektionen (schwere und langanhaltende Pyodermien, Dermatitiden im Nasen-Mund-Bereich, ekzematöse Läsionen, Lymphknotenvereiterungen und septische Bakterienabsiedelungen in Knochen, Darm, Leber und Lunge). Der Erkrankung liegt eine Mutation des HAXI(HCLS1-associated X1)-Gens zugrunde. Das mitochondriale HAXI-Protein schützt myeloische Zellen vor Apoptose. Defekte HAXI-Proteine resultieren in vermehrter Apoptose, was zur Reduktion der PMNs im Blut führt. Orale Manifestationen sind chronische Ulzerationen des harten Gaumens und der Alveolarmukosa, Gingivitiden und in der Pubertät beginnende, sehr rasch verlaufende Parodontitiden.
Die Therapie besteht aus einer lebenslangen Substitution mit gentechnisch hergestelltem Granulozyten-Wachstumsfaktor (G-CSF). Bei Patienten, die nicht auf G-CSF reagieren, bleibt nur die Knochenmarkstransplantation10.
Störungen des Zellstoffwechsels
Glykogenspeicher-Syndrome
Die Glykogenspeicher-Syndrome (GSD, Glykogenosen) sind autosomal-rezessiv vererbte Erkrankungen des Glykogenabbaus bzw. der Glykogensynthese mit pathologisch gesteigerter Glykogenspeicherung in vielen Organen (Leber, Nieren, Herz, Muskulatur, ZNS). Es gibt je nach vorhandenem Enzymdefekt verschiedene Formen bzw. Typen. GSD Typ 1b (ORPHA364/ICD-10: E74.0) wird durch einen Defekt der Glukose-6-Phosphat-Translokase verursacht.
Die Symptome von GSD 1b umfassen Hepatomegalie, Minderwuchs, Hypoglykämie, Neutropenie und Fehlfunktionen der PMNs. Eine pränatale Diagnostik ist nicht bei allen Formen möglich. Bei Glykogenspeicher-Syndrom 1b, das häufig mit einer Neutropenie einhergeht, werden auch starke Entzündungen der Gingiva und Parodontitis beschrieben11.
Hypophosphatasie
Die Hypophosphatasie (HPP; Phosphoethanolaminurie, Rathbun-Syndrom; ORPHA436/ICD-10: E83.30) ist eine eher seltene, vererbte Erkrankung, die mit Störungen der Knochenbildung (Kraniosynostose, Thoraxdeformitäten) und Zahnmineralisation einhergeht. Die Häufigkeit der schweren HPP-Formen wurde auf 1:100.000 Geburten geschätzt. Sie ist auf fehlende oder verringerte Aktivität der alkalischen Phosphatase (AP) im Serum und im Knochen zurückzuführen. Die Symptome sind je nach Erbgang und Mutation sehr variabel. Bisher wurden mehr als 190 Mutationen der nicht gewebespezifischen AP beschrieben.
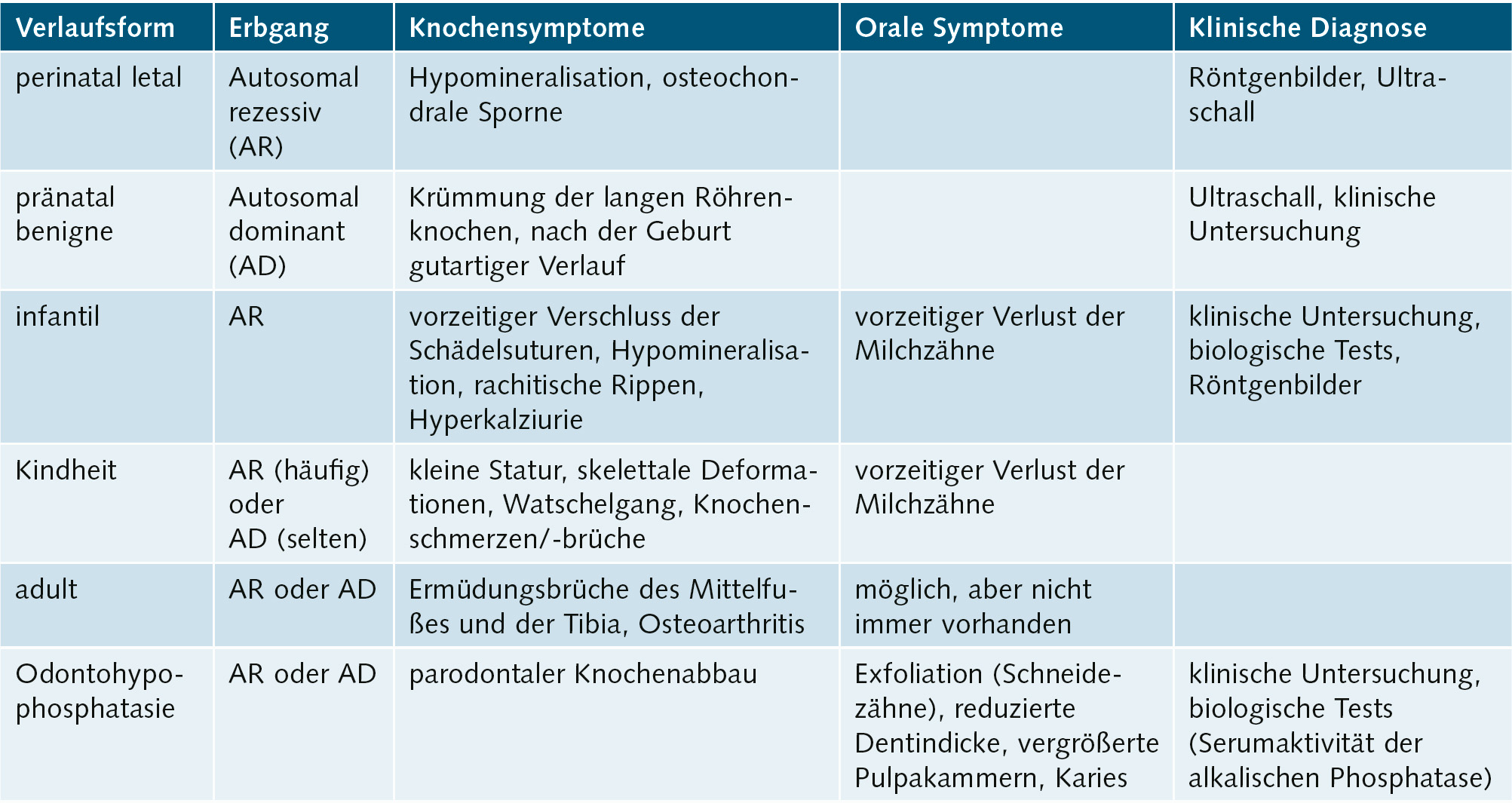
Symptomatik: Aktuell werden 6 verschiedene Formen unterschieden (Tab. 1). Die klinischen Ausprägungen reichen von der Todgeburt ohne Mineralisation der Knochen bis zu spät im Erwachsenenalter auftretenden Symptomen. Je ausgeprägter die Funktionsstörung der AP, desto schwerer verläuft die Hypophosphatasie. Während es bei der letalen perinatalen HPP zum Tod in utero oder nach wenigen Tagen zum Tod durch respiratorische Komplikationen kommt, tritt bei der benignen pränatalen HPP eine spontane Verbesserung der skelettalen Symptome ein. Patienten mit infantiler HPP erscheinen bei Geburt normal, entwickeln aber während der ersten 6 Lebensmonate die Symptome der HPP. Kleine Statur und vorzeitiger Milchzahnverlust sind häufig. Die KindheitsHPP ist durch eine lange Schädelform (Dolichocephalie) bedingt durch vorzeitige Mineralisation der Sagittalnaht, vergrößerte Gelenke, verspätetes Laufenlernen, kleine Statur und Watschelgang gekennzeichnet. Häufig treten Frakturen und Knochenschmerzen auf. Auch hier sind kleine Statur und vorzeitiger Milchzahnverlust, der zumeist bei den Schneidezähnen anfängt, häufig (Abb. 2). Sekundäre metabolische Entzündung in den Knochen und Hyperprostaglandinismus sind verbreitet. Die Erwachsenenform der HPP wird erst im mittleren Lebensalter symptomatisch. Häufig sind Fußschmerzen durch Ermüdungsbrüche des Metatarsus (Mittelfuß) oder Oberschenkelschmerzen durch Pseudofrakturen des Femur die ersten Beschwerden. Bei genauer Erhebung der zahnärztlichen Anamnese stößt man häufig auf vorzeitigen Verlust der Milchzähne. Die Odontohypophosphatasie ist durch vorzeitigen Verlust der Milchzähne bei komplettem Erhalt der Wurzel, großen Pulpakammern und starker Karies gekennzeichnet (Tab. 1). Die biochemischen Marker dieser Patienten unterscheiden sich nicht von denen der Kindheits- bzw. Erwachsenenform.
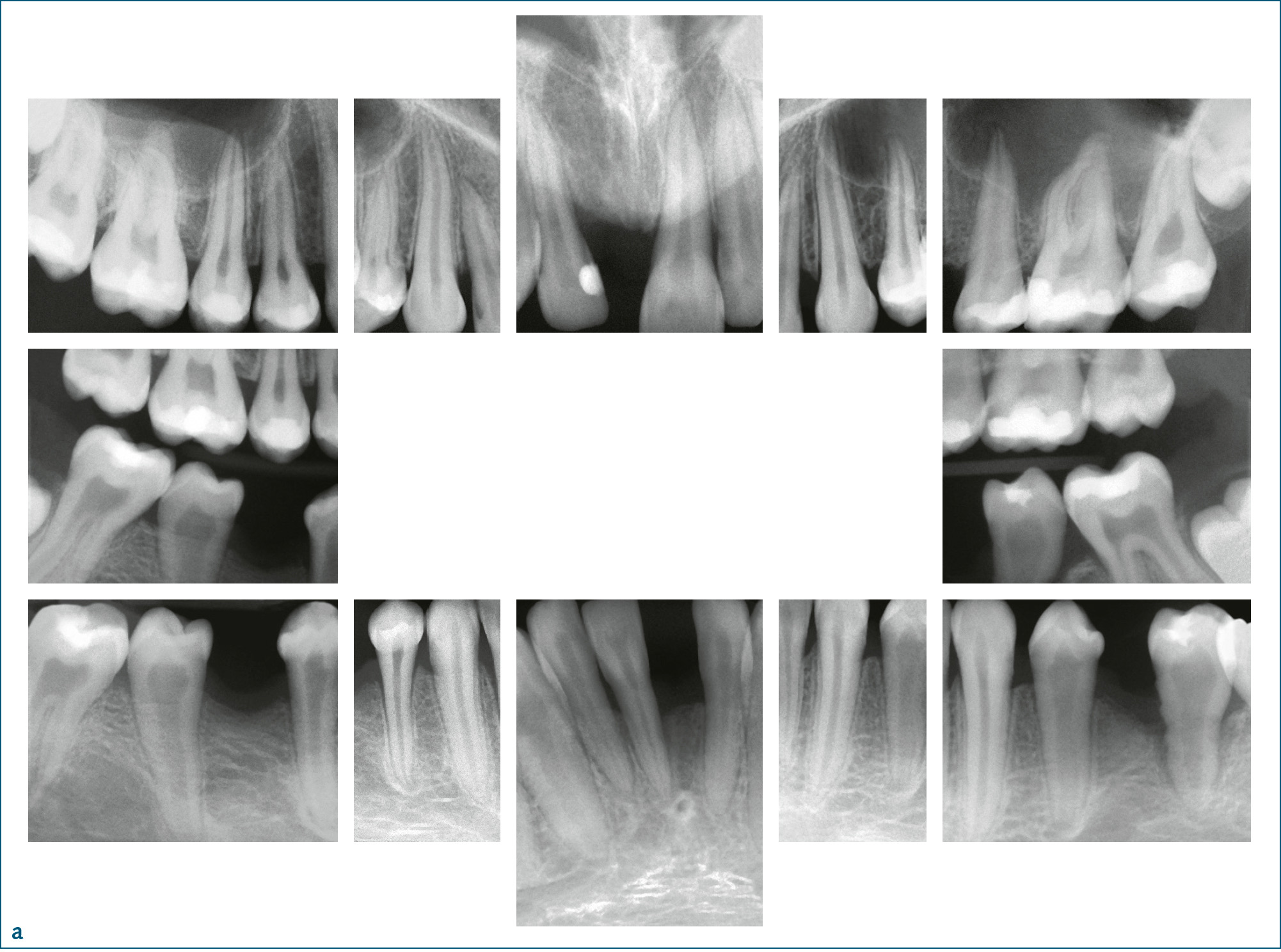
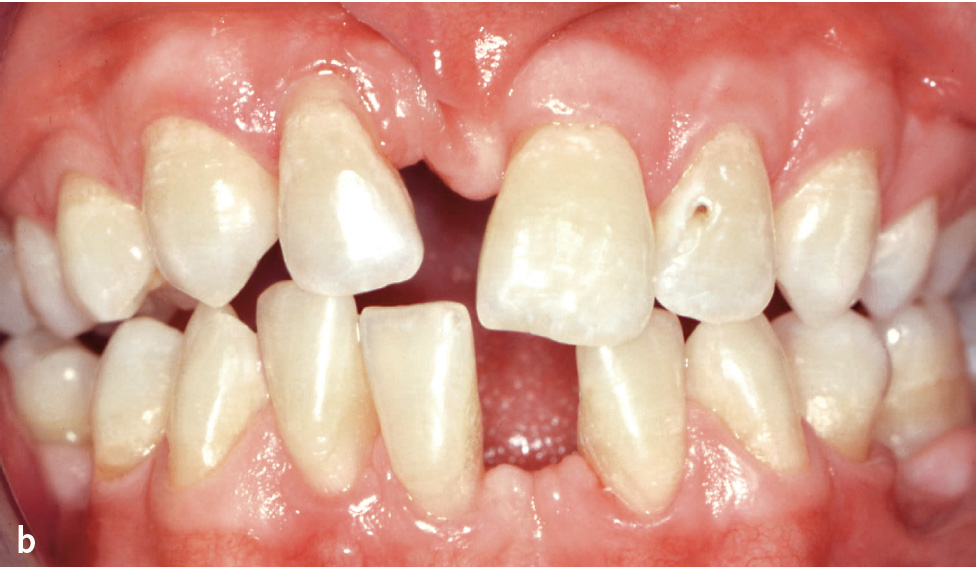
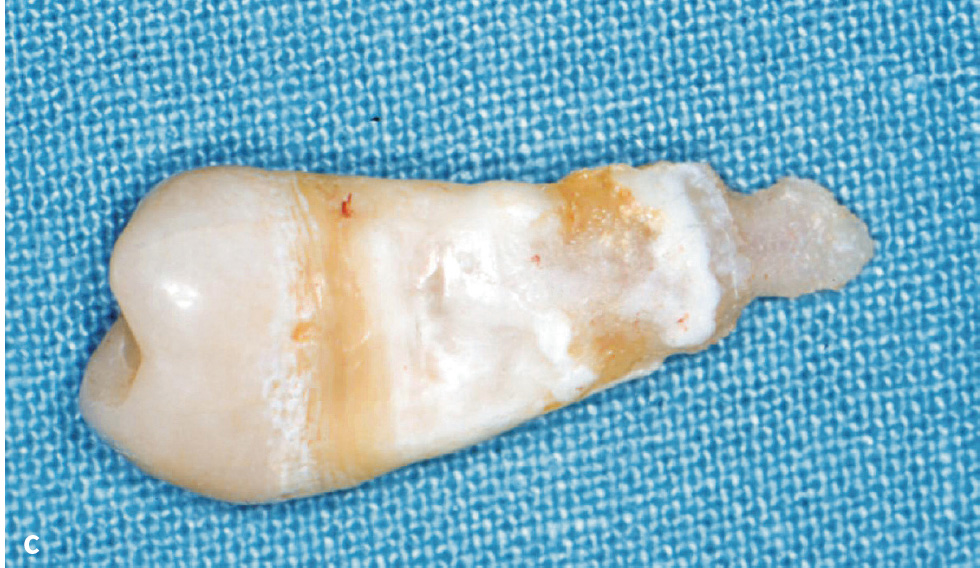
Abb. 2a bis c 14-jähriger männlicher Patient mit Kindheits-Hypophosphatasie. Die Beweglichkeit der Schultergelenke ist eingeschränkt und der Patient ist von kleiner Statur. Anamnestisch ergibt sich ein vorzeitiger Verlust der Milchzähne: a) Röntgenstatus: Die Zähne 11, 31 und 46 fehlen, es finden sich zahlreiche Restaurationen (Kariesvorgeschichte) und große Pulpakammern. Bei den Ober- und Unterkieferschneidezähnen und bei 35 zeigt sich Knochenabbau bis ins mittlere Wurzeldrittel. Die Wurzeloberfläche von 35 erscheint unregelmäßig; b) klinische Ansicht; c) Zahn 35 nach Extraktion aufgrund eines Parodontalabszesses: unregelmäßige, atypische Wurzeloberfläche.
Tab. 1 Klinische Verlaufsformen der Hypophosphatasie12.
Diagnostik: Neben den klinischen und röntgenologischen Verfahren tragen molekularbiologische Methoden zur Diagnose bei. Bei HPP ist die Aktivität der Serum-AP deutlich reduziert, wie aber auch z. B. in der frühen Schwangerschaft, bei Schilddrüsenunterfunktion, Anämie oder Zöliakie. Bei HPP ist Phosphoethanolamin im Urin erhöht. Ein sensitiver Marker für HPP ist ein erhöhter Pyridoxal-5‘-Phosphat-Spiegel12.
Ehlers-Danlos-Syndrom
Das Ehlers-Danlos-Syndrom (Fibrodysplasia elastica generalisata congenita; ORPHA98249/ ICD-10: Q79.6) ist eine Bezeichnung für eine Gruppe erblicher Krankheitsbilder mit Kollagendysplasie, die sich nach biochemischen, genetischen und klinischen Kriterien in zehn verschiedene Typen aufgliedert (Tab. 2). Die Ätiologie ist je nach Typ ein autosomal-dominanter, -rezessiver oder X-chromosomaler Erbmodus. Es gibt entsprechend unterschiedliche pathochemische Mechanismen der gestörten Kollagenfibrillogenese13.
Tab. 2 Formen des Ehlers-Danlos-Syndroms13.
TypErbgangBiochemieKlinische SymptomeIautosomal dominant (AD)unbekanntHyperelastizität und Wundheilungsstörungen der Haut, Überstreckbarkeit der GelenkeIIADunbekanntHyperelastizität und Wundheilungsstörungen der Haut, Überstreckbarkeit der GelenkeIIIADunbekanntausgeprägte Gelenkhypermobilität, wenig ausgeprägte HautsymptomatikIVAR (rezessiv) oder ADFehlen von Typ-III-KollagenRupturen im Bauchraum und der großen GefäßeVX-chromosomalunbekanntwie Typ II; stark dehnbare HautVIARDefekt der Lysyl-HydroxylaseAugenruptur, Kyphoskoliose, hyperelastische Haut, instabile GelenkeVIIADstrukturelle Mutation des Typ-I-Kollagenskongenitale Hüftendislokation, hypermobile Gelenke, dehnbare, purpurne HautVIII ParodontitisADunbekanntfragile Haut, prätibiale Vernarbung, ParodontitisIXX-chromosomaldefekte Kollagen-Kreuzverlinkungokzipitale Exostosen, Verbreiterung und Krümmung der Röhrenknochen an den Ansätzen von Bändern und Sehnen, deformierte Schlüsselbeine, milde Hautüberelastizität, verringerte Aktivität der Lysyl-OxidaseXARdefektes Plasma-FibronektinStriae, moderate Dehnbarkeit der Haut, Gelenkhypermobilität, ThrombozytenaggregationsdefektDie Symptome bestehen je nach Typ aus einer unterschiedlichen Symptomkonstellation und -schwere insbesondere mit Hyperelastizität und erhöhter Vulnerabilität sowie Wundheilungsstörungen der Haut und Überstreckbarkeit der Gelenke mit Luxationsneigung (Tab. 2). Insbesondere der Typ VIII manifestiert rasch verlaufende Parodontitiden. Allerdings wurde rasch verlaufende Parodontitis auch schon für Typ IV beschrieben13.