


- -
- 100%
- +
Recientemente, el Ministerio de Salud de Chile, en la última versión del Reglamento del Sistema Nacional de Control de los Productos Farmacéuticos de Uso Humano, publicada el 20 de diciembre del 2019, incluyó en el Decreto 50 la importación de productos farmacéuticos para uso exclusivo del importador (Minsal, 2019b). Dentro de las estrategias políticas se busca fomentar la importación con miras a disminuir el precio de los medicamentos (Miranda y Parada, 2019). En este marco regulatorio, el Instituto de Salud Pública (ISP) creó el Sistema Electrónico Importación Provisional y Clasificación de Mercancías (Sipro), el cual permite realizar solicitudes de importación de productos farmacéuticos, cosméticos y dispositivos médicos para uso individual (ISP, 2020). En suma, para su aprobación el isp solicita la presentación de los siguientes documentos (ISP, 2020): formulario correctamente diligenciado, documento de transporte, retención aduanera, receta médica, factura o boleta, poder y cédula de ciudadanía o pasaporte.
La solicitud se puede realizar por vía electrónica y de manera presencial en el isp, después de cancelarse la tarifa correspondiente.
Los medicamentos requeridos para tratar las patologías incluidas en la Ley Ricarte Soto los financia el sistema de salud chileno, mientras que los medicamentos importados para uso exclusivo del importador los asume el interesado y no son específicos para tratar enfermedades raras o huérfanas. Por este mecanismo es posible importar cualquier tipo de medicamento, siempre que sea autorizado por el ISP.
Colombia: medicamentos vitales no disponibles, Decreto 481 de 2004
En Colombia se adoptó la definición de enfermedad huérfana como aquella que es crónicamente debilitante, pone en peligro la vida y su prevalencia es menor a uno de cada 5000 personas (Arnold et al., 2015). Aunque no existe definición para medicamentos huérfanos, en el 2004 se emitió el Decreto 481 de 2004, “por el cual se dictan normas tendientes a incentivar la oferta de medicamentos vitales no disponibles en el país”. En esta disposición se establecen los criterios, las modalidades y los requisitos que permiten la importación y facilitan el acceso a estos medicamentos definidos como aquellos indispensables e irremplazables para un paciente o grupo de pacientes.
La Sala Especializada de Medicamentos de la Comisión Revisora10 del Instituto Nacional de Medicamentos y Alimentos (Invima) define y actualiza periódicamente un listado de medicamentos que deben estar en normas farmacológicas11 y cumplir con los tres criterios estipulados en el artículo 4 del Decreto 481 de 2004:
• que no se encuentre en fase de investigación;
• que no se comercialice en el país o, en su defecto, se encuentre en condición de desabastecimiento;
• que no cuente con alternativas disponibles en el país.
De igual manera, en el decreto se determinó que, para los medicamentos vitales no disponibles, se concede la exención del registro sanitario y se permite la importación bajo tres modalidades diferentes: para paciente específico, uso exclusivo en casos de urgencia clínica y para más de un paciente. Los requisitos en cada uno de los tipos de solicitudes se describen en la tabla 2.
TABLA 2. Requisitos para importación de medicamentos vitales no disponibles
TIPO DE SOLICITUDREQUISITOSArtículo 8: autorización de importación para un paciente especifico• Solicitud expresa. Carta en la que se manifiesta la intención de importación del medicamento y se describe la información general de la solicitud. • Copia del documento de identidad del paciente. • Principio activo, concentración y presentación. • Fórmula médica y resumen de la historia clínica, en la que se indica la dosis, el tiempo de duración del tratamiento y el nombre del medicamento. La fórmula debe estar firmada por el médico tratante, con la indicación y el número de la tarjeta profesional. • Copia del recibo de consignación acorde a la tarifa establecida por el Invima.Artículo 9: autorización de importación de medicamentos vitales no disponibles para uso exclusivo en casos de urgencia clínica• Bastará la sustentación médica del medicamento solicitado. • Copia del recibo de consignación acorde a la tarifa establecida por el Invima.Artículo 10: autorización de importación para más de un paciente de medicamentos vitales no disponibles• Solicitud expresa. Carta donde se manifiesta la intención de importación del medicamento; se describe la información general de la solicitud. • Certificado de venta libre (CVL) o certificado de producto farmacéutico. • Certificado de existencia y representación legal del solicitante. • Certificado de análisis. • Copia del recibo de consignación acorde a la tarifa establecida por el Invima.Fuente: Decreto 481, 2004.
Aunque, según el artículo 9, los requisitos sean menores en las solicitudes presentadas para uso exclusivo en casos de urgencia, en la práctica estos requerimientos se presentan cumpliendo los requisitos establecidos en el artículo 8 del mismo decreto. Además, a diferencia de las solicitudes de autorización de importación para un paciente, los requerimientos para más de un paciente no puede radicarlos una persona natural; en este caso, lo podrá realizar cualquier entidad pública o privada que se encuentre legalmente constituida, en atención a los requisitos dispuestos en el artículo 10.
En el 2013 el Invima emitió la circular dg-100-00022-13, “Autorización de importación de medicamentos vitales no disponibles para más de un paciente”. En esta se aclara cuál es el propósito de las solicitudes para más de un paciente y se resalta que el Invima puede ejercer acciones de inspección, vigilancia y control, por lo que los importadores deben conservar la información y documentación sobre la importación, la comercialización y la distribución de los medicamentos vitales no disponibles, tales como:
• listado de pacientes o criterios identificadores del grupo de pacientes;
• ciudad en que se ubican los pacientes;
• instituciones prestadoras de servicios de salud (IPS) que atienden los pacientes;
• datos de médico tratante;
• demás información que permita la trazabilidad.
En esta línea de discusión, a partir del año 2016 el Invima solicita como requisito adicional en las tres modalidades, el identificador único del medicamento (IUM) reglamentado en la Resolución 3166 de 2015, “por medio de la cual se define y se implementa el estándar de datos para medicamentos de uso humano en Colombia”. En complemento, el 29 de diciembre del 2018, el Ministerio de Salud y Protección Social y el Ministerio de Comercio, Industria y Turismo emitieron el Decreto 2498 de 2018, “por el cual se determina la permanencia de un reglamento técnico en materia de medicamentos vitales no disponibles en el país”. De esta manera, la publicación se realiza en atención al artículo 2.2.1.7.6.7 del Decreto 1074 de 2015, “por medio del cual se expide el Decreto Único Reglamentario del Sector Comercio, Industria y Turismo”. En este documento se exige una revisión por parte de la entidad regulatoria de las normas emitidas, con el fin de determinar su permanencia, modificación o derogatoria, al menos una vez cada cinco años, y así verificar si existe algún cambio conforme a las causas que dieron origen a la norma (Decreto 1074, 2015).
En este decreto también se determina la permanencia del reglamento técnico para el Decreto 481 de 2004, lo cual denota que no se requiere ningún tipo de modificación porque la salud pública del país, en lo que concierne a la importación y comercialización de medicamentos vitales no disponibles, no ha cambiado en catorce años o, sencillamente, se advierte que no se dio el espacio propicio para revisar la norma con los diferentes actores involucrados.
Aunque en el decreto señalado no se estipula qué dependencia debe realizar el estudio de las solicitudes de medicamentos vitales no disponibles, es necesario señalar que de acuerdo con el numeral 5 del artículo 23 del Decreto 2078 de 2012, “por el cual se establece la estructura del Instituto Nacional de Vigilancia de Medicamentos y Alimentos-Invima y se determinan las funciones de sus dependencias”, la Dirección de Operaciones Sanitarias es la encargada de tramitar, estudiar y emitir el concepto sanitario sobre licencias y autorizaciones de importación, esto en conformidad con la normatividad vigente. Por tanto, estas solicitudes son radicadas ante el Invima y estudiadas por el grupo de autorizaciones y licencias para importación y exportación de la Dirección de Operaciones Sanitarias.
Una vez se reciben las solicitudes de autorización de importaciones, el profesional a cargo del estudio del trámite determina si esta cumple con los requisitos establecidos en el Decreto 481 de 2004, acorde al tipo de petición presentada por el interesado y, de modo simultáneo, se verifica si el medicamento está incluido en el listado de medicamentos vitales no disponibles12. De ser así, el trámite únicamente lo estudia la Dirección de Operaciones, y en caso contrario, esta dependencia requerirá pronunciamiento de la Dirección de Medicamentos y Productos Biológicos. Luego del análisis, al margen de los dos casos, el grupo de autorizaciones determina si la solicitud se puede autorizar, negar o si se necesita información adicional para continuar con el estudio (Invima, 2018). En consecuencia, a corte del 2019 había un total de 249 medicamentos incluidos en el listado de medicamentos vitales no disponibles.
En el caso de que una solicitud requiera información adicional, el solicitante cuenta con treinta días calendario, prorrogables, para presentar la respuesta ante la dependencia encargada del estudio, según lo establecido en el Código de Procedimiento Administrativo y de lo Contencioso Administrativo (Ley 1437, 2011). Así, el solicitante puede o no presentar respuesta satisfactoria y, dependiendo de esto, se determina si se autoriza o se niega la solicitud. Es importante indicar que en ocasiones los interesados desisten o abandonan su pretensión. En estos casos se genera una resolución de negación por desistimiento expreso o tácito según corresponda (Invima, 2018).
En el diagrama de flujo de la figura 1 se detalla el proceso de autorización de importación de medicamentos vitales no disponibles.
FIGURA 1. Solicitudes de medicamentos vitales no disponibles
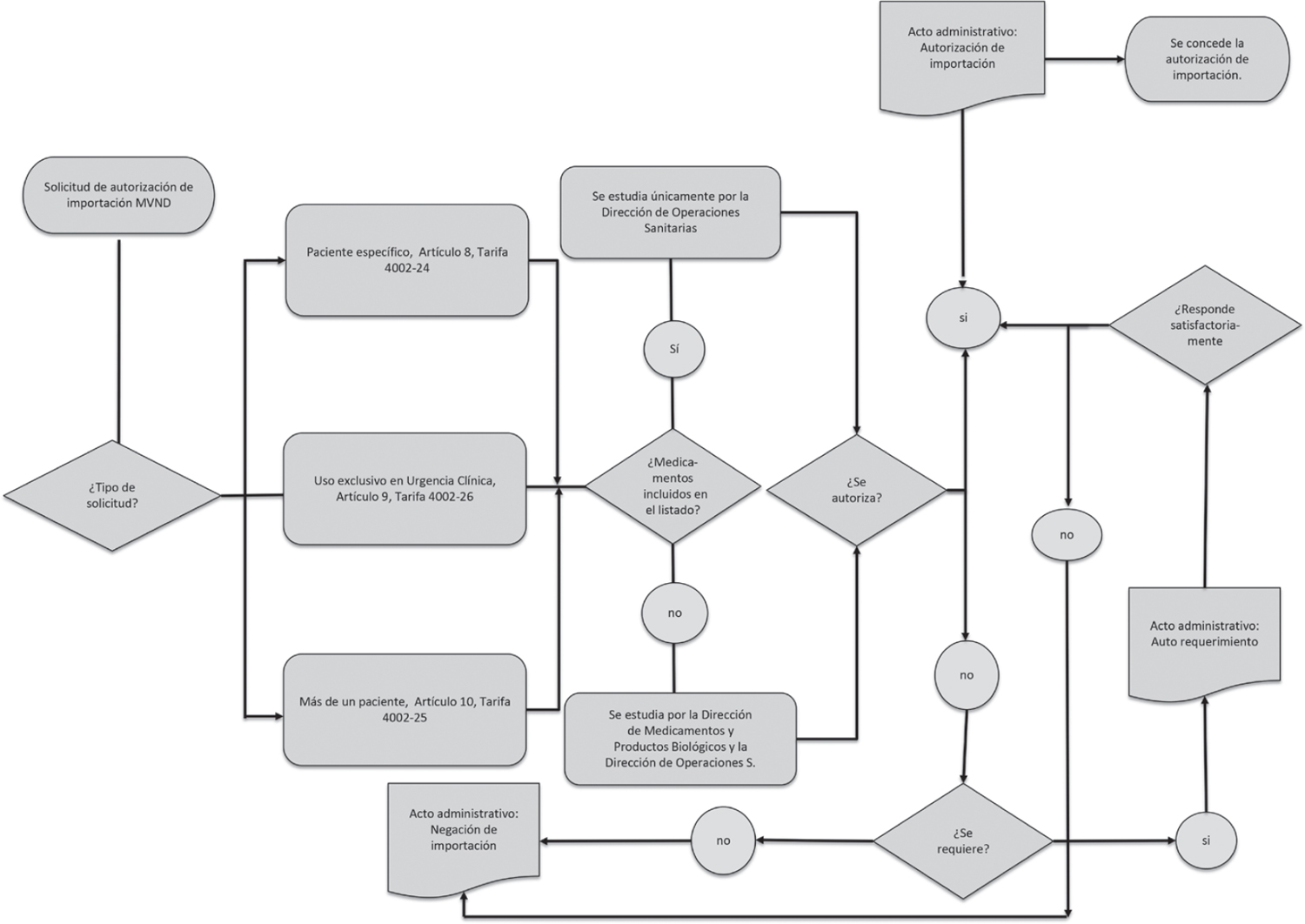
Fuente: elaboración propia.
Ahora bien, las autorizaciones concedidas deben ser presentadas ante la ventanilla única de comercio exterior (VUCE). El interesado debe cumplir diligenciando la licencia de importación acorde con lo establecido en la guía de diligenciamiento de intenciones de importación ante la VUCE, disponible en la página web del Invima (2019).
Según lo estipulado por el artículo 15 de la Ley Estatutaria de Salud (Ley 1751, 2015), el sistema de salud no cubre tecnologías en salud si no existe evidencia científica acerca de su seguridad y eficacia clínica, o sobre su efectividad clínica. No obstante, el parágrafo tercero del mismo artículo establece: “Bajo ninguna circunstancia deberá entenderse que los criterios de exclusión definidos en el presente artículo, afectarán el acceso a tratamientos a las personas que sufren enfermedades raras o huérfanas”. Aunque algunos medicamentos para enfermedades raras se encuentran incluidos en el Plan de Beneficios con cargo a la unidad de pago por capitación (UPC)13, en el caso en el que el paciente requiera un medicamento no incluido, el médico debe hacer la prescripción por el sistema Mipres14 (Minsalud, 2017). Además, en caso de ser autorizado el medicamento o haberse obtenido por orden judicial, lo financia la Administradora de los Recursos del Sistema General de Seguridad Social en Salud (Ley 1955, 2019). En lo que respecta al régimen de exceptuados y/o especiales (Fuerzas Armadas, universidades, magisterio y demás) y quienes no hacen parte del sgsss, la financiación viene de los recursos de cada régimen (Vargas-Peláez et al., 2019).
Análisis comparativo de las legislaciones sobre medicamentos huérfanos o vitales no disponibles de los países incluidos en el estudio
Una vez revisadas las normas a nivel internacional y nacional, en la tabla 3 se establecen las diferencias y semejanzas que existen en la legislación colombiana con respecto a las estructuradas en las agencias sanitarias mencionadas (EMA, AEMPS, FDA, Cofrepris, ANMAT, ISP, Invima).
TABLA 3. Comparación de la normativa internacional y la colombiana sobre medicamentos para enfermedades huérfanas
PAÍSUNIÓN EUROPEAESPAÑAESTADOS UNIDOSMÉXICOARGENTINACHILECOLOMBIAAGENCIA SANITARIAAgencia Europea de Medicamentos (EMA)Agencia Española de Medicamentos y Productos Sanitarios (AEMPS)Food and Drugs Administration (FDA)Comisión Federal para Protección Contra Riesgos Sanitarios-(Cofepris)Administración Nacional de Medicamentos, Alimentos y Tecnologías- (ANMAT)Instituto de Salud Pública (ISP)Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima)AÑO DE EMISIÓN20002009198320122012/20172016 /20192004NORMAReglamento (CE) no 141/2000Real Decreto 1015Orphan Drug Act (public law 07-414)Por el que se adicionan los artículos 224 BIS y 224 BIS 1 a la Ley General de SaludDisposición 4622/Disposición di-2017-10874Ley Ricarte Soto; Ley 20.850 Decreto 50Decreto 481TÉRMINOSMedicamentos huérfanosMedicamentos huérfanos; medicamentos en situaciones especiales (medicamentos utilizados en investigación o uso compasivo, medicamentos extranjeros, medicamentos off label).Medicamentos huérfanosMedicamentos huérfanosEspecialidades medicinales cuyo registro será concedido “bajo condiciones especiales/medicamentos que no cuentan con registro en el país”.Tecnologías sanitarias para enfermedades o patologías de alto costo/importación de medicamentos para uso exclusivo del importador.Medicamentos vitales no disponiblesFINALIDADEstablecer un procedimiento comunitario para declarar determinados medicamentos como medicamentos huérfanos y establecer incentivos para fomentar la investigación, el desarrollo y la comercialización de los medicamentos declarados huérfanos.Reducir la carga burocrática para el solicitante y la agencia durante la solicitud de medicamentos no autorizados o no comercializados a nivel nacional (Real Decreto 1015, 2009).Establecer los fundamentos legislativos para incentivar el desarrollo de medicamentos para enfermedades y afecciones poco conocidas, denominados como medicamentos huérfanos, a través de la reducción de costos y los estímulos financieros en la producción y el desarrollo.Implantar medidas y acciones para impulsar y fomentar la disponibilidad de medicamentos huérfanos, a través de medicamentos más asequibles, mediante el fomento de la investigación y el desarrollo.Establecer un procedimiento operativo para tramitar solicitudes de especialidades medicinales o medicamentos destinados a la prevención, el diagnóstico y/o el tratamiento de enfermedades poco frecuentes, o enfermedades sin tratamientos disponibles a nivel nacional.Crear un sistema de protección financiera para diagnóstico y tratamiento de alto costo/facilitar el acceso.Determinar la base normativa para la identificación de los medicamentos vitales no disponibles y establecer los incentivos para la investigación, el desarrollo, la producción, la importación y la comercialización de los medicamentos mencionados.INCENTIVOS O FLEXIBILIDADESExclusividad de mercado por diez años; protocolo de asistencia y seguimiento; tarifas regulatorias reducidas.Se acoge a la EMA.Exclusividad de mercado por siete años; protocolo de asistencia y seguimiento; tarifas regulatorias; crédito fiscal en ensayos clínicos.Exención registro sanitario.Exención registro sanitario.N/AExención de registro sanitario.FINANCIACIÓN EN EL SISTEMA DE SALUDDesignación de huérfanos a nivel europeo y autorización de comercialización y reembolsos se realizan a nivel nacional.Exige un proceso de fijación de precios y financiación, pero no cobija a todos los medicamentos; unos los asume el sistema y otros no (gastos de bolsillo).Dependiendo del tipo de modelo de seguridad, este lo asume el seguro médico o el paciente (gasto de bolsillo).Falta de regulación, por lo que en su gran mayoría son asumidos por gastos de bolsillo.Reembolso condicionado al resultado del tratamiento. En algunos casos gasto de bolsillo.Los medicamentos establecidos en los protocolos de las dieciocho patologías son asumidos 100 % por el sistema de salud.Son asumidos por el sistema de saludLISTADO DE MEDICAMENTOS AUTORIZADOS ACTUALIZADO A 20191654 medicamentosNo refiere5223 medicamentos89 medicamentos reconocidos al año 2018No refiere27 patologías y sus protocolos249 medicamentosFuente: elaboración propia
Diferencias en la fecha de emisión
La primera diferencia son las fechas de emisión de las legislaciones relacionadas con los medicamentos huérfanos, objeto de este trabajo. Estados Unidos reconoce hace 37 años a los medicamentos huérfanos, seguido por la Unión Europea con veinte años. Mientras que en América Latina, en los países estudiados en esta investigación, aparece Colombia como pionera.
A pesar de no contemplar en su legislación el término medicamentos huérfanos, Colombia tuvo la iniciativa de regular los medicamentos vitales no disponibles, dentro de los cuales se incluyen aquellos indicados en el tratamiento de enfermedades raras desde el año 2004. México, Argentina y Chile establecieron sus regulaciones solo en la segunda década del siglo XXI. Esta diferencia temporal puede estar relacionada con el hecho de que los mercados farmacéuticos de los países no despiertan el mismo interés de la industria para la comercialización de sus productos, por tanto, la presión tecnológica que sufren los sistemas de salud para cubrir estas tecnologías, mediante las prácticas de marketing de la industria farmacéutica, no se produce al mismo tiempo (Ramírez y Oliver, 2018; Vargas-Peláez et al., 2014; Vargas-Peláez et al., 2019).
Otros factores que pueden explicar esta diferencia temporal son el fortalecimiento y la mayor visibilidad de las organizaciones de pacientes, algunas veces financiadas por la industria farmacéutica; un mayor reconocimiento de los pacientes de la garantía del acceso a este tipo de medicamentos como garantía de su derecho fundamental a la salud por parte de los Estados, y la acción del poder judicial para atender las demandas de estos pacientes, fenómeno que se ha documentado en los países latinoamericanos incluidos en este estudio y que ha tenido un mayor desarrollo en la década de 2010 (Ramírez y Oliver, 2018; Vargas-Peláez et al., 2014; Vargas-Peláez et al., 2019).
Diferencias en el término establecido
La segunda diferencia entre las regulaciones es el término establecido en cada país. Entre estos se encuentran “medicamentos huérfanos”, “medicamentos en situaciones o condiciones especiales”, “tecnologías en salud para enfermedades” o “patologías de alto costo” y “medicamentos vitales no disponibles”. Se destaca que en el caso de Colombia el término es más amplio, pues abarca no solo aquellos que se requieren para patologías raras, sino que también incluye medicamentos que no se encuentran disponibles por desabastecimiento. Lo mismo se observa en países como España, México, Argentina y Chile, cuya regulación, además de otorgar reconocimiento de medicamentos huérfanos o necesarios para tratar enfermedades poco frecuentes dentro del territorio nacional, también ofrecen mecanismos que permiten la importación de medicamentos no disponibles en ese país, promoviendo así el acceso a estos.
En lo que concierne a España, los autores García y Alonso indicaron cómo con el Real Decreto 1015/2009 se proporcionó en este país una amplia cobertura legal a los medicamentos no legislados hasta ese momento, tales como las preparaciones galénicas de productos que no se comercializan por falta de interés comercial (García y Alonso, 2010).
Diferencias en la finalidad normativa
Por otra parte, en el caso de la finalidad de cada normatividad, se observa que mientras en Estados Unidos y la Unión Europea el principal objetivo es fomentar la investigación y el desarrollo sobre enfermedades raras, en las regulaciones de España, México, Colombia, Chile y Argentina el principal objetivo es facilitar los trámites para la importación de los productos, teniendo como incentivo o flexibilidad la exención de registro sanitario. Aunque la regulación mexicana y colombiana mencionan que entre sus fines está el fomento de la investigación y del desarrollo, en estos países no se realiza este tipo de actividades, pues normalmente se llevan a cabo en países desarrollados (p. ej. la UE y E. E. U. U.) (Schuhmacher et al., 2016). En el caso de Chile se destaca la Ley Ricarte Soto, cuyo principal objetivo es la cobertura de los tratamientos de algunas enfermedades huérfanas por parte del sistema de salud.
Financiación
La financiación de los medicamentos huérfanos ha sido un desafío importante para los sistemas de salud debido a sus altos costos, así como por la incertidumbre en torno a su eficacia y seguridad, pues los ensayos clínicos usualmente son de corto espectro. A la fecha, ningún sistema de salud ha tenido la capacidad de asumir el 100 % de la financiación de todos los medicamentos huérfanos (Stawowczyk et al., 2019; Mayrides et al., 2020; Hughes-Wilson et al., 2012). En los países latinoamericanos, aunque algunos sistemas cubren medicamentos específicos, en el caso en que el medicamento no esté incluido, el paciente puede recurrir al poder judicial para obtener acceso (Stawowczyk et al., 2019; Mayrides et al., 2020; Hughes-Wilson et al., 2012).
Listados de medicamentos
En relación con la publicación de medicamentos aprobados como huérfanos o medicamentos vitales no disponibles, Estados Unidos lidera la cantidad autorizada; lo siguen la Unión Europea, Colombia y México. En Argentina no hay una publicación o un listado en los que se indique los medicamentos autorizados o aprobados como huérfanos. Chile, por su parte, tiene un listado en el que se señalan las enfermedades con sus respectivos protocolos de atención, además de los medicamentos que se requiere para tratar la patología.
Por otro lado, la diferencia en la cantidad de aprobaciones entre Estados Unidos y la Unión Europea puede ser el resultado de aspectos tales como los criterios para definir si una enfermedad es huérfana (si es por prevalencia o por número de pacientes en el país), pero también por la rigurosidad del examen de las solicitudes. En el segundo caso, se puede citar el ejemplo de la designación por parte de la FDA del remdesivir como medicamento huérfano en marzo del 2020. El remdesivir es un fármaco que actualmente puede ser alternativa de tratamiento para la infección por Covid-19. La decisión se basó en que en el momento de la solicitud de Gilead (laboratorio productor), esta enfermedad afectaba a menos de 200.000 personas en Estados Unidos. No obstante, debido a la presión mediática y social, Gilead desistió de esta designación, la cual en caso de comprobarse que el remdesivir es efectivo para el Covid-19, habría generado grandes barreras de acceso (Fang y Lerner, 2020; Mancini et al., 2020; Owens, 2020).
Características de los requisitos
Algunas similitudes encontradas entre las regulaciones se refieren al requisito de contar con el consentimiento informado firmado por el paciente para tramitar la solicitud de importación en España, Argentina y Chile, así como la posibilidad que existe de tramitar solicitudes de medicamentos que se requieren con urgencia en Argentina y Colombia. Estos aspectos reflejan la preocupación de los reguladores frente a la satisfacción de la necesidad en salud de los pacientes, y que cuenten con la información necesaria sobre los riesgos del tratamiento. De esta manera, la responsabilidad es compartida entre el médico prescriptor, el paciente y la entidad sanitara, la cual es la que finalmente autoriza el trámite.
Por último, llama la atención que solo en la regulación de Argentina se contempla la necesidad de que el paciente sea vinculado a un programa de farmacovigilancia, de modo que se pueda hacer seguimiento a los resultados del tratamiento, así como la necesidad de que el médico presente una declaración de conflicto de interés en cuanto mecanismo para controlar los efectos del marketing farmacéutico sobre los hábitos de prescripción que han sido documentados (Jacob, 2018), de modo que se garantice que la utilización del medicamento corresponda a la necesidad del paciente.